2022. 8. 25. 01:55ㆍ게임만 하니 공부도 해야지
안녕하세요!
1편에 이어서 돌아온 LAMMPS 2편입니다!
이번 시간에는 간단한 시뮬레이션을 돌려보고 자신이 돌리는 시뮬레이션을 직접 볼수 있는 프로그램을
소개해 드리겠습니다.
2편을 보기 전에 1편부터 보고오세요!
Lammps 기본 사용법 정리 1편 (MD 시뮬레이션)
안녕하세요 이번 시간에는 간단하게 기본적인 코드에 대해 알아보고 실제로 돌려보도록 하겠습니다!! 일단 기본적인 설치 및 실행은 구글에 lammps라고 검색하면 공식 사이트가 나오는데 거기서
dalgon-game.tistory.com
units metal
boundary p p p
atom_style charge
read_data sio2.data
1편에서 이렇게 끝났죠?
복습해 보면
units metal 은 사용할 단위
boundary p p p 은 반복을 설정해주고
atom_style charge 은 사용할 원자의 형태
read_data sio2.data 은 사용할 시뮬레이션의 크기와 각 원자의 위치정보입니다.
여기까지가 저번편 정리구요
이렇게 되면 시뮬레이션에 원자가 위치해만 있는 상태입니다
저희가 원하는 건 실제로 어떻게 움직이는지 보고싶은 것이니깐
몇가지 설정을 해 주어야 합니다
설정해주어야 하는 것은 간단한데요.
크게 두가지 입니다!
첫번째!
Force Field입니다. (포텐셜)
혹시 모르시면 이 글을 참고해주세요!
LAMMPS의 근간이자 MD 시뮬레이션의 기초이므로 꼭 아셔야 합니다
LAMMPS 정보 공유 시작합니다(1) LAMMPS 란?
요즘 한참 리뷰를 안썼는데 다시 돌아왔습니다 이번에는 제가 대학원에서 공부하는 프로그램에 대한 정보를 알려드리려고 합니다 제 글이 100퍼센트 맞는 것은 아니고 제가 순수하게 맨땅에 헤
dalgon-game.tistory.com
최대한 간단하게 설명하고 넘어갔으나실제로는 LJ 포텐셜 하나만 사용하는 것이 아니라
사용하는 물질의 상이나
원자인지 분자인지
어떤 물질인지에 따라
다른 포텐셜을 사용합니다.
예를 들어, 합금의 경우 2nn MEAM
이온결합이나 공유결합의 경우 COMB , ReaxFF
등등이 있어 필요시 다른 포텐셜을 사용합니다.

간단한 리뷰글이니 추천드립니다!
저희의 경우
SiO2를 사용하죠?
원래 실리콘 다이옥사이드에 저는 pedone 포텐셜을 사용하지만
사용 방법이 까다롭기 때문에
이번에는 LJ 포텐셜을 사용하겠습니다!
★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆
이때 사용하는 물질에 무슨 포텐셜이 필요한지는 관련 논문을 찾으셔야 합니다!
★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆★☆
그럼 LAMMPS 매뉴얼로 가보죠!
5.8. Pair_style potentials — LAMMPS documentation
© Copyright 2003-2022 Sandia Corporation.
docs.lammps.org
여기로 가 보면 pair_style 이 보이실 겁니다
들어가보면

엄청나게 많은 포텐셜들이 있을겁니다...
당연히 적용할 수 있는 포텐셜이 많은 프로그램이 좋은 프로그램이므로 많습니다 ㅋㅋㅋ
물론 없는 포텐셜도 적용 가능하긴 한데 그건 나중시간에~
여기서
여기 있는 포텐셜이 전부 LJ 포텐셜을 기반으로 하는 포텐셜입니다
저희가 사용할 것은 가장 기초인
를 사용합니다!
내부 문서를 확인하면
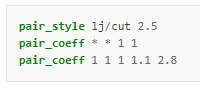
라고 적혀있고
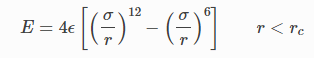
사용 포텐셜의 식

필요 매개변수
이렇게 적혀있네요
해석해보면

이렇습니다
그럼 저 매개변수는 어디서 찾느냐!
논문에서 찾으면 되겠죠?
적당한 것을 찾아보니

amorphous SiO2 인 것은 아쉽지만
어짜피 저희도 이후에 녹일 것이기 때문에 그냥 사용해 보겠습니다.
논문을 보다 보면
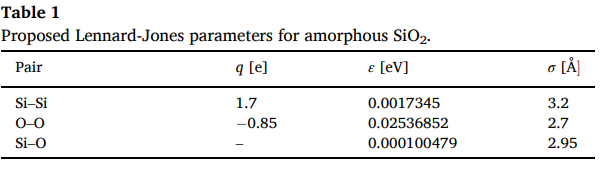
테이블과 논문에서 사용한 식이 있겠죠?
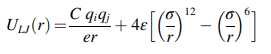
근데 식을 보면 저희가 사용한 식과 약간 다릅니다
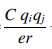
이게 추가되었는데 이는 전하에 따른 힘
즉 Coulombic pairwise interaction 입니다.
그래서 저희는
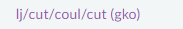
lj/cut에 coul이 추가된 pair/style을 사용합니다!
사용법은 위와 동일하며 매개변수를 위에 테이블에서 가져오면
인풋파일에
pair_style lj/cut/coul/cut 10
Si - Si
pair_coeff 1 1 0.0017345 3.2
Si - O
pair_coeff 1 2 0.000100479 2.95
O - O
pair_coeff 2 2 0.02536852 2.7
이렇게 적으면 됩니다!
제가 뒤에 컷오프를 생략한 이유는 어짜피 pair_style lj/cut/coul/cut 10
여기서 10으로 설정했기 때문에 따로 설정해주지는 않았습니다.
또한 전하도 설정해줘야 하니 위에 테이블에서 보고
Si charge
set type 1 charge 1.7
O charge
set type 2 charge -0.85
설정해줍니다.
1 과 2가 뭔지는 sio2.data 파일에 나와있습니다!
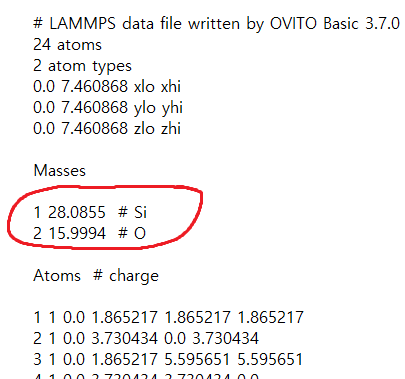
여기서 1이 Si, 2가 O 입니다.
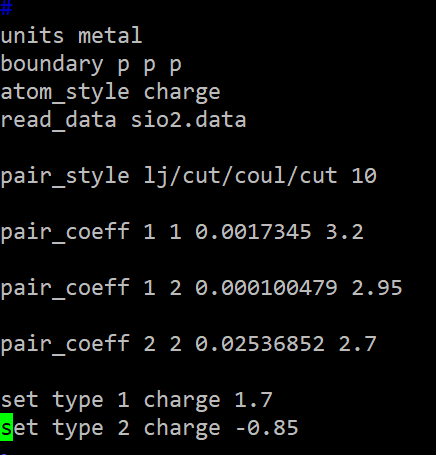
이렇게 인풋파일에 넣으면
포텐셜이 적용됩니다!
두번째!
Fix 커맨드입니다!
Fix 커맨드는 다음 시간에 더 자세히 알려드리구 일단 기본적인것만 알려드리겠습니다!
Fix 커맨드 또한 카테고리 커맨드에 있습니다!
Fix 커맨드는 우리가 원하는 시뮬레이션의 상세 설정을 하는 역할로
종류 또한 엄청나게 많습니다.
가장 기본적인 Fix 커맨드는 세가지로
NVE : 가장 기초적인 앙상블로 추가 설정이 없음
NVT : 온도 설정 가능
NPT : 압력 설정 가능 및 온도 설정 가능
저희는 오늘은 원자가 어떻게 움직이는지 보고 싶기 때문에
가장 기본적인 nve를 사용합니다
fix 1 all nve
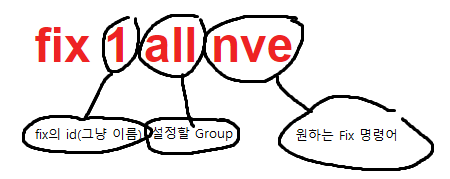
마지막으로 몇 step이나 돌릴것인지!
run 10000
이 숫자도 원하시는대로 설정하시면 됩니다!
마지막으로 눈으로 보고싶으니
dump 1 all atom 100 SiO2.dump

이렇게 하면
최종 인풋 파일은
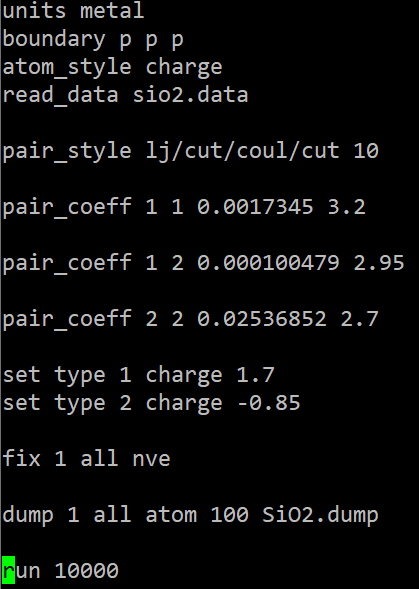
이렇게 됩니다!
이러고 파일을 돌리면!
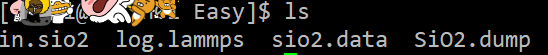
dump 파일이 생성되었죠?
저 파일을 OVITO에 넣으면
이렇게 진동하는 모습을 볼 수 있습니다!!!!!!!!!!
드디어 저희는 간단한 시뮬레이션을 돌릴 수 있게 되었습니다!
하지만 의문점이 있죠
초반부에 움직이지 않는 모습을 보여주는데 그 이유가 있습니다
그 이유와 오늘 간단하게 넘어간 Fix , run , dump 외 다른 명령어에 대한 설명 및
더 큰 시뮬레이션은 다음 시간에 설명하겠습니다!
궁금한점 있으시면 댓글 남겨주세요!
Lammps 기본 사용법 정리 3편 (MD 시뮬레이션)
Lammps 기본 사용법 정리 2편 (MD 시뮬레이션) 안녕하세요! 1편에 이어서 돌아온 LAMMPS 2편입니다! 이번 시간에는 간단한 시뮬레이션을 돌려보고 자신이 돌리는 시뮬레이션을 직접 볼수 있는 프로그
dalgon-game.tistory.com
'게임만 하니 공부도 해야지' 카테고리의 다른 글
| Lammps 기본 사용법 정리 4편 (MD 시뮬레이션) 로그 보는법 (8) | 2022.12.05 |
|---|---|
| Lammps 기본 사용법 정리 3편 (MD 시뮬레이션) (10) | 2022.11.22 |
| VASP, DFT 시뮬레이션 사용법 공략 1편 (6) | 2022.07.20 |
| Lammps 기본 사용법 정리 1편 (MD 시뮬레이션) (12) | 2022.07.09 |
| LAMMPS 정보 공유 시작합니다(1) LAMMPS 란? (0) | 2022.06.26 |