2022. 7. 20. 23:12ㆍ게임만 하니 공부도 해야지
안녕하세요!
저번엔 LAMMPS 사용법을 알려드렸는데요! 곧 2편도 올라갈 예정입니다
이번엔 LAMMPS와는 다른!
시뮬레이션 프로그램 사용법을 알려드리겠습니다
제가 알려드릴 VASP는 MD 시뮬레이션이 아닌 DFT
즉, Density Funtional Theory 밀도 함수 이론 방법입니다
말이 굉장히 어려운데 간단하게 설명해보자면
MD에서는 원자를 간단히 공으로만 생각했다면!
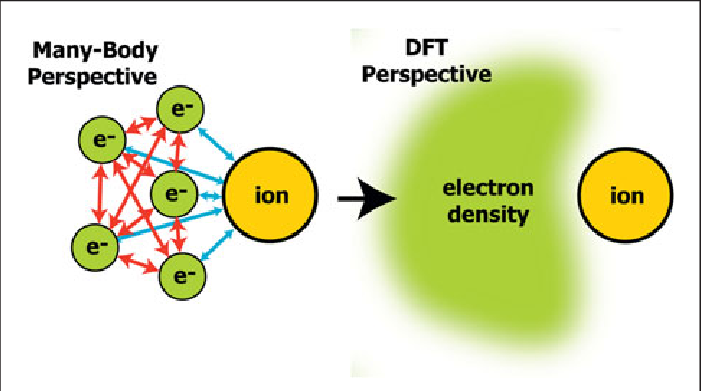
DFT에서는 공이 아닌 전자와 이온사이의 작용까지 계산한다고 볼 수 있습니다.
계산이 상당히 까다롭고 오랜 시간이 걸리지만 MD보다 훠어어어어얼씬 정확합니다
실제 원자의 움직임과 거의 동일하다고 볼 수 있죠
DFT에서는 전자까지 계산한다고 했죠!
하지만 실제로 전자 하나 하나를 계산하는 것은 아닙니다...
하나하나의 계산은 실제로는 불가능하죠
그래서 저희는 전자를 하나하나보지 않고 하나의 밀도로 봅니다.
그럼 파동함수로 표현이 가능하고

슈뢰딩거 방정식을 계산할 수 있습니다
물론 바닥 상태의 에너지만요
DFT는 방법론을 사용하기 때문에 계산이 반복적이고 그로인하여 시간이 오래 걸립니다.
그래서 MD처럼 10000개~1000000개 정도의 원자를 시뮬레이션 하면 아마 인류가 멸망할 때쯤 끝날 수도 있겠네요...
그래서 DFT로는 적은 개수의 원자를 사용해 MD에서는 불가능한 정밀한 계산을 합니다.
예를 들면 결합 에너지나 cohesive E, surface E 등등 거의 모든 계산을 할 수 있고 LAMMPS 보다
더 거칠지만 많은 정보를 포함하므로 더 잘 사용할수록 무궁무진한 정보를 얻을 수 있습니다.
VASP는 그 중에 아마 DFT에서 가장 유명한 프로그램이고 가장 빠른 프로그램이라고 볼 수 있습니다.
가격도 굉장히 비싸죠... 교수님 왈 약 1억 8천만원정도..?
저는 학교 라이센스를 사용해 사용이 가능합니다...
https://www.vasp.at/wiki/index.php/The_VASP_Manual
The VASP Manual - Vaspwiki
Getting started Featured topics Category subtopics (amongst others) Theoretical background Density-functional theory, projector-augmented-wave method, molecular dynamics, GW approximation, etc. Calculation setup Installation, input files, output files, INC
www.vasp.at
VASP 또한 램스처럼 나름? 친절한 메뉴얼 사이트가 있지만
사용 방법은 지옥과 같습니다...
다음 시간에는 인풋 파일과 실행법에 대해 알려드리겠습니다!!
다음 시간에 뵈요!
'게임만 하니 공부도 해야지' 카테고리의 다른 글
| Lammps 기본 사용법 정리 4편 (MD 시뮬레이션) 로그 보는법 (8) | 2022.12.05 |
|---|---|
| Lammps 기본 사용법 정리 3편 (MD 시뮬레이션) (10) | 2022.11.22 |
| Lammps 기본 사용법 정리 2편 (MD 시뮬레이션) (21) | 2022.08.25 |
| Lammps 기본 사용법 정리 1편 (MD 시뮬레이션) (11) | 2022.07.09 |
| LAMMPS 정보 공유 시작합니다(1) LAMMPS 란? (0) | 2022.06.26 |