2022. 12. 5. 23:48ㆍ게임만 하니 공부도 해야지
생각보다 많은 분들이 Lammps 즉 MD 시뮬레이션에 흥미가 있으신 것 같아
다음편을 내봅니다
제가 저번편에서 Fix 명령어에 대해 설명했죠?
Fix 명령어는 각각 사용하는 때마다 다른 Fix 명령어를 사용하기 때문에
모든 걸 설명해드릴 수가 없어요 ㅠㅠㅠ 대신 모르는게 있으시면 댓글 남겨주시고
LAMMPS Mailing List Mirror
This category is a read-only mirror of the LAMMPS mailing list, made available for historical context and searching. New posts and questions should be made to the parent category.
matsci.org
저는 모르는 게 있으면 이 사이트를 참고합니다!
일단 이전편을 보고 와주세요!
Lammps 기본 사용법 정리 3편 (MD 시뮬레이션)
Lammps 기본 사용법 정리 2편 (MD 시뮬레이션) 안녕하세요! 1편에 이어서 돌아온 LAMMPS 2편입니다! 이번 시간에는 간단한 시뮬레이션을 돌려보고 자신이 돌리는 시뮬레이션을 직접 볼수 있는 프로그
dalgon-game.tistory.com
이번 시간에는 log 파일을 보는법을 알려드리겠습니다!
요건 저번에 보여드린 시뮬레이션을 돌린 곳인데요!
여기 보시면 log.lammps 라는 파일이 만들어졌죠?
이는 제가 돌린 시뮬레이션의 로그가 들어있는 파일입니다
내부를 보시면
(이 로그는 제 시뮬레이션이므로 이전에 돌린 시뮬레이션하고 다르니 예시로 보여드립니다.)
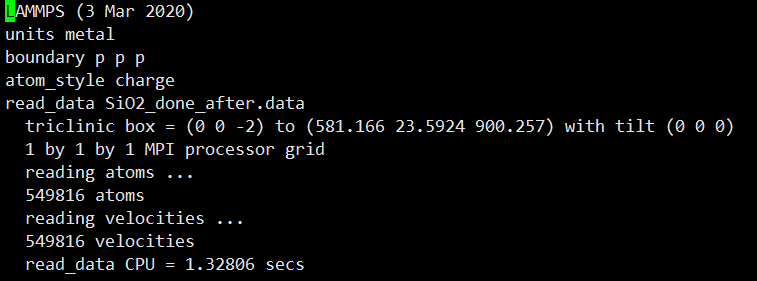
위에서부터 제가 설정한
단위
바운더리
원자 종류
read_data 한 데이터파일명
그 밑에 triclinic box 부터는 시뮬레이션 박스의 크기입니다.
각각 x축 0~581
y축 0~ 23.59
z축 -2 ~ 900
tilt는 어느정도 휘어졌는지 입니다.
이 정보는 저번에 알려드렸다시피 read_data 에서 불러온 것이므로 제가 설정한 파일에 들어있는 정보를 불러온 것입니다.
그 밑에 1 by 1 by 1은 사용하는 mpi의 수 즉 코어를 몇개 사용하는지의 정보입니다.
이 시뮬레이션은 1개의 코어를 썻지만 주로 저는 대량의 원자를 사용하므로 40개 정도 사용합니다.
아마 윈도우에서 사용하는 lammps는 1개코어만 가능할텐데 (확실하지 않음)
저는 리눅스를 사용합니다!
밑에 뭐 몇개 원자 몇개 속도
얼마나 걸렸는지 등등 잡정보도 많습니다.
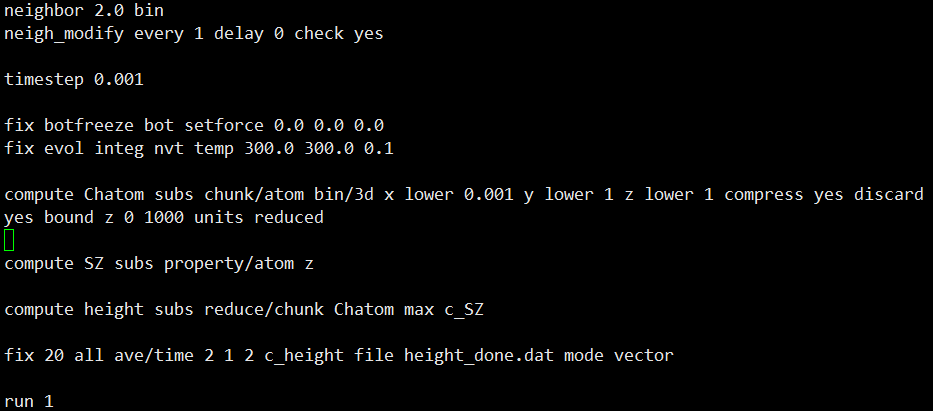
이 밑에는 제가 설정한 명령어들이 존재합니다
보시면 처음보는 compute 명령어도 있고
저번에 알려드린 fix 명령어도 있죠?
compute 명령어는 다음에 알려드리겠습니다!
맨 마지막 run 이후에는 설정한 fix 대로 시뮬레이션이 진행됩니다.
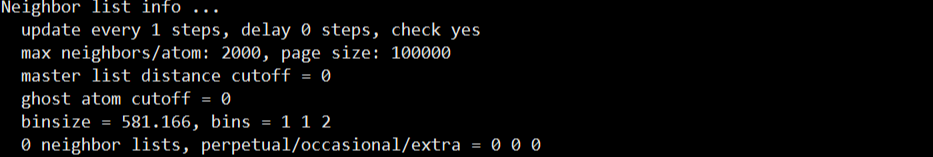
맨 첫번째로 나오는 Neighbor list info ...
밑에 6번째 줄 까지는 Neighbor 의 정보입니다.
이는 명령어
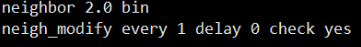
를 통해 설정한 내용인데요
제가 lammps에 대해 설명할 때 매 스텝마다 각각의 힘을 계산해서 움직인다고 했죠?
하지만 사실은 그렇지 않습니다.
아니 그럴수도 있고요
제가 위에서 timestep 을 0.001 로 설정했죠?
units metal의 시간 단위는 ps입니다
제 시뮬레이션에서 0.001ps 니까 제 timestep은 fs 입니다
사실 fs는 굉장히 짧은 시간입니다. 그러므로 lammps에서 대규모 시뮬레이션을 할때
neigh_modify delay 10 을 주게 되면 1번 계산하고 이 힘을 가지고 10step 동안 움직이게 됩니다.
어 그러면 정확하지 않지 않나요? 왜 그러나요?
맞습니다 정확하지 않게 됩니다. 하지만 그만큼 계산량이 1/10배로 떨어지고 계산속도가 빨라지죠?
또한 떨어진 정확도도 시뮬레이션의 크기가 크고 원자 수가 굉~~~장히 많아지면 티도 나지 않게 됩니다.
이는 전체적인 정보 즉 부피나 밀도, 압력같은 정확성이 크지 않아도 되는 작업에 쓰고
저는 실제적인 수치가 중요한 시뮬레이션을 하기 때문에 delay를 0을 주고 매 순간마다 계산하게끔
every 를 1로 주었습니다.
정확한 neigh_modify는
neighbor command — LAMMPS documentation
This command sets parameters that affect the building of pairwise neighbor lists. All atom pairs within a neighbor cutoff distance equal to the their force cutoff plus the skin distance are stored in the list. Typically, the larger the skin distance, the l
docs.lammps.org
역시 매뉴얼을 참고해주세요!
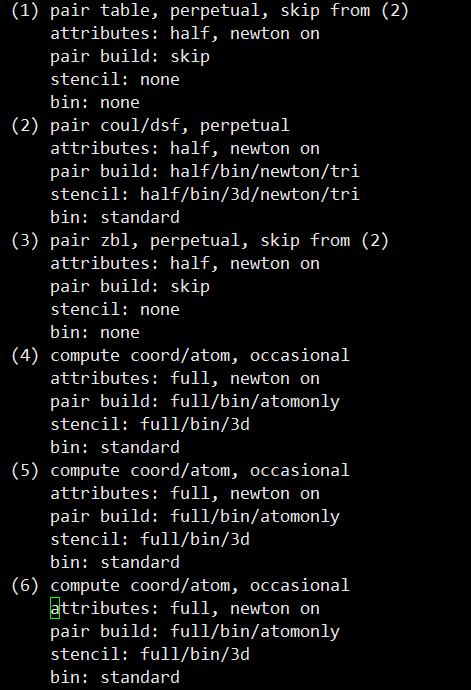
그 다음엔 이 시뮬레이션에 설정된 Pair_style
즉 적용된 원자 사이의 규칙을 알려주고
설정한 compute 명령어를 보여줍니다.
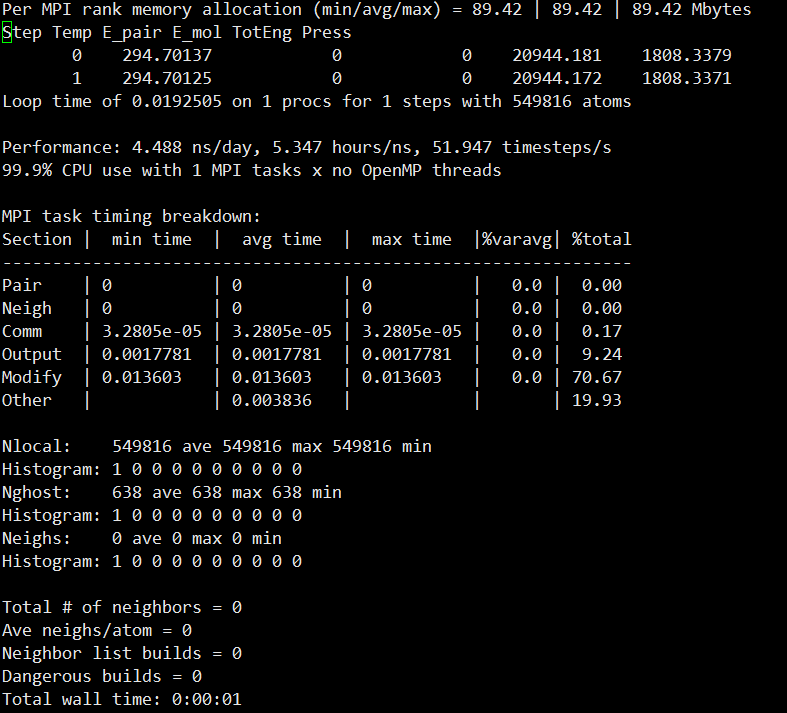
이제 중요한 부분인데 저기 2번째줄에
Step Temp E_pair E_mol TotEng Press
이렇게 보이시나요?
저 순서대로 밑에 숫자들의 순서입니다.
스탭 온도 pair_에너지 분자에너지 총_에너지 압력입니다.
이는 설정한 앙상블에 따라 다르게 나오고
thermo 명령어에 따라 몇 step마다 나오는지
thermo_style custom을 사용하면 나오는 정보를 다르게 설정할 수 있습니다.
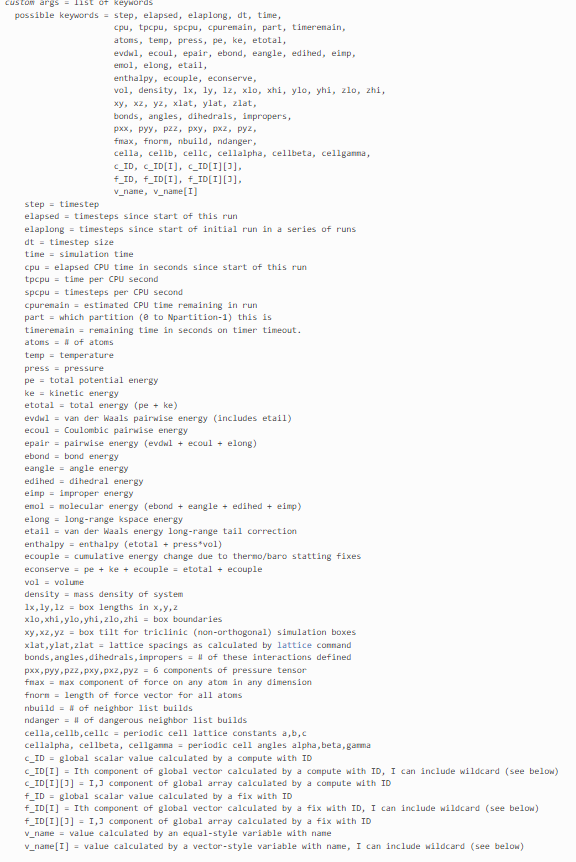
무려 이게 전부 커스텀 키워드로 출력 가능한 정보들입니다.
필요한 정보를 thermo_style custom으로 뽑아오면 log 파일에 설정한 step마다 나오겠죠?
하지만 원하는 정보가 저기 키워드에 없을 수도 있잖아요??
그럼 어떡하죠... 연구중단?
그런 상황에서 필요한 것이 compute 명령어 입니다.
compute는 다음 시간에 알려드리도록 하겠습니다!
★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★
로그파일은 굉장히 중요합니다.
여러분이 시뮬레이션을 돌리다 오류가 나면 그 이유가 뭔지 어디서 멈췄는지
알 수 있고 또 thermo를 통해 원하는 정보를 뽑아낼 수도 있습니다.
★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★★
아무튼
lammps 처음해 보시면 오류가 굉~~~~~~~~~~~~~~~~~~~~~~~~장히 많이 날 텐데
그때마다 로그 보시고 오류를 수정하시면 되겠습니다.
매우 친절한 우리 lammps는 오류가 왜 생겼는지 뭐가 문제인지 다 알려주므로 잘 하실겁니다.
뭐든 시행착오는 성공의 어머니입니다...
실패를 두려워하지 마시고 뭐든 하고싶은게 있으면 돌려봅시다!
다음시간에 만나용~
궁금한게 있으시면 언제든 댓글 남겨주세요
저도 모르는게 있으니 함께 알아가요!
'게임만 하니 공부도 해야지' 카테고리의 다른 글
| Lammps 기본 사용법 정리 5편 (MD 시뮬레이션) Compute & Fix 명령어 사용법 (4) | 2023.07.23 |
|---|---|
| Lammps 기본 사용법 정리 3편 (MD 시뮬레이션) (10) | 2022.11.22 |
| Lammps 기본 사용법 정리 2편 (MD 시뮬레이션) (21) | 2022.08.25 |
| VASP, DFT 시뮬레이션 사용법 공략 1편 (6) | 2022.07.20 |
| Lammps 기본 사용법 정리 1편 (MD 시뮬레이션) (11) | 2022.07.09 |